























[대학저널 온종림 기자] 서울대학교 보건대학원 및 생물정보협동과정 성주헌 교수와 생물정보협동과정 박사과정 박건형 연구원이 유전체 분석을 혁신할 수 있는 새로운 원천 알고리즘을 개발하여 공개했다. 유전체 분석은 질병 진단과 신약 개발 등의 첨단 바이오 테크놀로지에 필수적인 핵심 기술이다.
유전체 분석은 실험적인 분석 과정뿐 아니라, 그 이후에 대규모의 생물정보 데이터 처리 단계가 필수적이다.
한 사람당 30억개의 염기서열을 분석하는 유전체 자료는PB (페타바이트) 규모에 달하여, 정확한 염기쌍을 효율적이고 빠르게 찾는 알고리즘은 매우 중요한 원천기술이다. 지금까지 이러한 원천 알고리즘은 전적으로 영-미의 기술에 의존해 왔다.
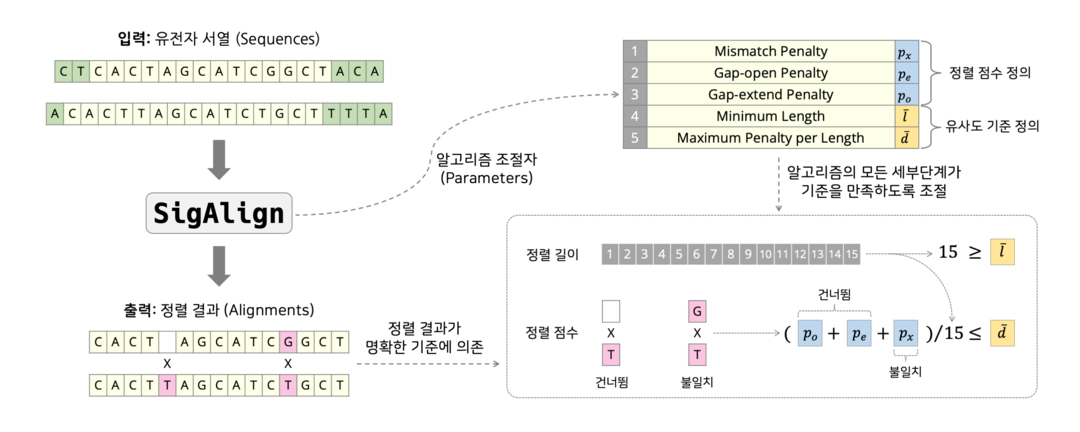
서울대 연구진이 개발한 “SigAlign” (Similarity-guided Alignment)은 현재 널리 사용되고 있는 알고리즘 (BWA-MEM, HISAT2, Bowtie2) 등에 비해 수십 배 빠른 속도로 더 정확한 결과를 도출하였으며, 표준 컴퓨팅 환경에서의 방대한 비교분석에서 초당 10만 개 이상의 데이터를 처리할 수 있는 유일한 알고리즘이었다.
또 SigAlign은 기존 알고리즘들이 수십 개의 난해한 설정값 (parameter) 옵션들을 조정해야 되고, 이런 조정과정을 거쳐도 결과를 예측하기 어려운 단점을 획기적으로 극복해, 5개의 순수한 생물학적 설정값 (불일치 점수를 제외하면 단 2개의 설정값) 만으로 유전체 분석의 모든 과정과 결과를 투명하고 예측 가능하게 만들었다는 점에서 큰 관심을 받고 있다.
연구진이 개발한 유전체분석 알고리즘의 정확성과 투명성 및 직관적인 사용편의성 등은 유전체 분석이 필요한 모든 분야의 분석을 최적화 시킬 수 있는 방법이 될 것으로 예상된다.
[저작권자ⓒ 대학저널. 무단전재-재배포 금지]


















